Relaxation time prediction for a light switchable peptide by molecular dynamics
21-Jun-2010
Phys. Chem. Chem. Phys., 2010, 12, 6204-6218 published on 21.06.2010
Phys. Chem. Chem. Phys.
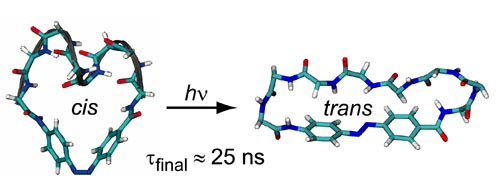
We study a monocyclic peptide called cAPB, whose conformations are light switchable due to the covalent integration of an azobenzene dye. Molecular dynamics (MD) simulations using the CHARMM22 force field and its CMAP extension serve us to sample the two distinct conformational ensembles of cAPB, which belong to the cis and trans isomers of the dye, at room temperature. For gaining sufficient statistics we apply a novel replica exchange technique. We find that the well-known NMR distance restraints are much better described by CMAP than by CHARMM22. In cAPB, the ultrafast cis/trans photoisomerization of the dye elicits a relaxation dynamics of the peptide backbone. Experimentally, we probe this relaxation at picosecond time resolution by IR spectroscopy in the amide I range up to 3 ns after the UV/vis pump flash. We interpret the spectroscopically identified decay kinetics using ensembles of non-equilibrium MD simulations, which provide kinetic data on conformational transitions well matching the observed kinetics. Whereas spectroscopy solely indicates that the relaxation toward the equilibrium trans ensemble is by no means complete after 3 ns, the 20 ns MD simulations of the process predict, independently of the applied force field, that the final relaxation into the trans-ensemble proceeds on a time scale of 23 ns. Overall our explicit solvent simulations cover more than 6 micros.