
Research Area C
Publications 2008
18-Feb-2011
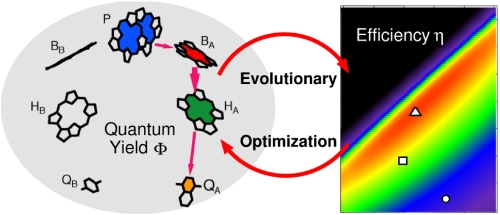
Photochemical solar energy conversion is considered as an alternative of clean energy. For future light converting nano-machines photosynthetic reaction centers are used as prototypes optimized during evolution. We introduce a reaction scheme for global optimization and simulate the ultrafast charge separation in photochemical energy conversion. Multiple molecular ... READ MORE
01-Apr-2009
The radiationless decay mechanisms of the S1 excited states of the 7H-keto-amino, 7H-enol-amino, and 7Hketo- imino tautomers of guanine have been investigated with the CASPT2//CASSCF method. Out-of-plane deformation of the six-membered ring or the imino group as well as dissociation of NH bonds have been considered as photochemical pathways leading to conical ... READ MORE
01-Apr-2009
We present the excited-state potential energy profiles of the biologically relevant 9H-keto-amino tautomer of guanine with respect to the radiationless decay via the out-of-plane deformation of the six-membered ring as well as the dissociation of NH bonds. The CASPT2//CASSCF method is employed for the reaction-path calculations. The reaction path for the out-of-plane ... READ MORE
19-Mar-2009
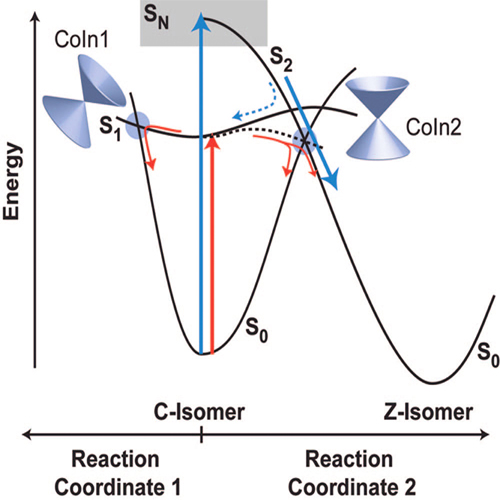
The photoinduced electrocyclic ring-opening of a fluorinated indolylfulgide is investigated by stationary and ultrafast spectroscopy in the UV/vis spectral range. Photoreactions, initiated by optical excitation into the S1 (570 nm) and SN (340 nm) absorption band of the closed isomer, lead to considerable differences in reaction dynamics and quantum yields. ... READ MORE
11-Apr-2008
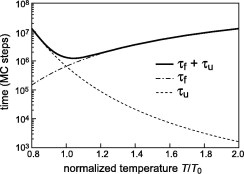
Replica exchange (RE) molecular dynamics (MD) simulations are frequently applied to sample the folding–unfolding equilibria of β-hairpin peptides in solution, because efficiency gains are expected from this technique. Using a three-state Markov model featuring key aspects of β-hairpin folding we show that RE simulations can be less efficient than conventional ... READ MORE
22-Feb-2008
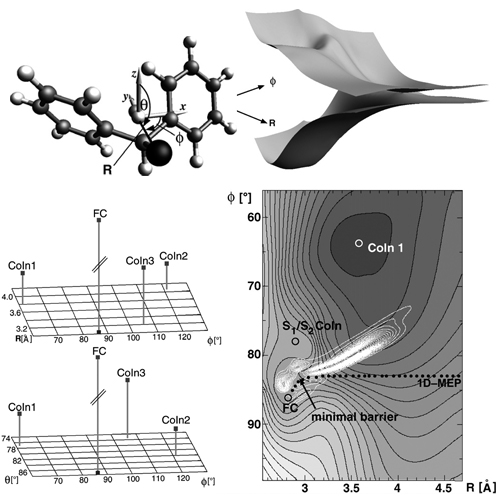
The primary processes in the formation of electrophilic precursor ions are studied on a microscopic scale by quantum chemical and quantum dynamical methods. For the competing reaction channels of heterolysis and homolysis in the photochemically induced dissociation of diphenylmethyl chloride, ab initio data for the ground and excited electronic states are ... READ MORE
17-Jan-2008
Molecular dynamics (MD) simulations of bulk liquid water at different thermodynamic conditions or of biomolecules in aqueous solution require a molecular mechanics (MM) force field that accounts for the sizable electronic polarizability α of the water molecule. A considerable number of such polarizable water models has been suggested in the past. Most of them ... READ MORE
The temperature steers the equilibrium and nonequilibrium conformational dynamics of macromolecules in solution. Therefore, corresponding molecular dynamics simulations require a strategy for temperature control which should guarantee that the experimental statistical ensemble is also sampled in silico. Several algorithms for temperature control have been proposed. ... READ MORE

